Simplifying Genomic Annotations in R
In this blog post I will be introducing cellbaseR R package, which makes use of the exhaustive RESTful Web service API that has been implemented for the Cellabase database. It enable researchers to query and obtain a wealth of biological information from a single database saving a lot of time. Another benefit is that researchers can easily make queries about different biological topics and link all this information together as all information is integrated
# to get the default CellbaseR object (human data, from genome GRCh37)
library(cellbaseR)
# A default cellbaseR object is created as follows
cb <- CellBaseR()
The CellbaseR Methods
The cellbaseR package provide many methods to query the cellbase webservices, they include:
- getGene
- getSnp
- getProtein
- getTranscript
- getRegion
- getVariant
- getClinical
- getTf
- getXref
In all cases the user is expected to provide the ids for the query and the resource to be queried, in addition to the CellbaseQuery object they created. For example, a query through the cbGene will look something like this
getGene
library(cellbaseR)
cb <- CellBaseR()
genes <- c("TP73","TET1")
res <- getGene(object = cb, ids = genes, resource = "info")
str(res,2)
## 'data.frame': 2 obs. of 12 variables:
## $ id : chr "ENSG00000078900" "ENSG00000138336"
## $ name : chr "TP73" "TET1"
## $ biotype : chr "protein_coding" "protein_coding"
## $ status : chr "KNOWN" "KNOWN"
## $ chromosome : chr "1" "10"
## $ start : int 3569084 70320413
## $ end : int 3652765 70454239
## $ strand : chr "+" "+"
## $ source : chr "Ensembl" "Ensembl"
## $ description: chr "tumor protein p73 [Source:HGNC Symbol;Acc:HGNC:12003]" "tet methylcytosine dioxygenase 1 [Source:HGNC Symbol;Acc:HGNC:29484]"
## $ transcripts:List of 2
## ..$ :'data.frame': 14 obs. of 20 variables:
## ..$ :'data.frame': 1 obs. of 20 variables:
## $ annotation :'data.frame': 2 obs. of 2 variables:
## ..$ expression:List of 2
## ..$ diseases :List of 2
# as you can see the res dataframe also contains a transcripts column
# which is in fact a list column of nested dataframes, to get the
# trasnscripts data.frame for first gene
TET1_transcripts <- res$transcripts[[1]]
str(TET1_transcripts,1)
## 'data.frame': 14 obs. of 20 variables:
## $ id : chr "ENST00000378295" "ENST00000604074" "ENST00000354437" "ENST00000357733" ...
## $ name : chr "TP73-001" "TP73-006" "TP73-002" "TP73-202" ...
## $ biotype : chr "protein_coding" "protein_coding" "protein_coding" "protein_coding" ...
## $ status : chr "KNOWN" "KNOWN" "KNOWN" "KNOWN" ...
## $ chromosome : chr "1" "1" "1" "1" ...
## $ start : int 3569084 3569084 3569129 3569129 3569129 3598930 3598930 3607236 3607236 3607236 ...
## $ end : int 3652765 3652765 3649856 3650467 3650467 3649643 3649643 3649856 3649856 3652765 ...
## $ strand : chr "+" "+" "+" "+" ...
## $ genomicCodingStart: int 3598930 3598930 3598930 3598930 3598930 3598930 3598930 3607470 3607470 3607470 ...
## $ genomicCodingEnd : int 3649643 3649326 3649326 3649643 3649643 3649643 3649643 3649326 3648119 3649643 ...
## $ cdnaCodingStart : int 156 156 111 111 111 1 1 235 235 235 ...
## $ cdnaCodingEnd : int 2066 1367 1610 1778 1733 1668 1623 1587 1515 1998 ...
## $ cdsLength : int 1910 1211 1499 1667 1622 1667 1622 1352 1280 1763 ...
## $ proteinID : chr "ENSP00000367545" "ENSP00000475143" "ENSP00000346423" "ENSP00000350366" ...
## $ proteinSequence : chr "MAQSTATSPDGGTTFEHLWSSLEPDSTYFDLPQSSRGNNEVVGGTDSSMDVFHLEGMTTSVMAQFNLLSSTMDQMSSRAASASPYTPEHAASVPTHSPYAQPSSTFDTMSP"| __truncated__ "MAQSTATSPDGGTTFEHLWSSLEPDSTYFDLPQSSRGNNEVVGGTDSSMDVFHLEGMTTSVMAQFNLLSSTMDQMSSRAASASPYTPEHAASVPTHSPYAQPSSTFDTMSP"| __truncated__ "MAQSTATSPDGGTTFEHLWSSLEPDSTYFDLPQSSRGNNEVVGGTDSSMDVFHLEGMTTSVMAQFNLLSSTMDQMSSRAASASPYTPEHAASVPTHSPYAQPSSTFDTMSP"| __truncated__ "MAQSTATSPDGGTTFEHLWSSLEPDSTYFDLPQSSRGNNEVVGGTDSSMDVFHLEGMTTSVMAQFNLLSSTMDQMSSRAASASPYTPEHAASVPTHSPYAQPSSTFDTMSP"| __truncated__ ...
## $ cDnaSequence : chr "TGCCTCCCCGCCCGCGCACCCGCCCGGAGGCTCGCGCGCCCGCGAAGGGGACGCAGCGAAACCGGGGCCCGCGCCAGGCCAGCCGGGACGGACGCCGATGCCCGGGGCTGC"| __truncated__ "TGCCTCCCCGCCCGCGCACCCGCCCGGAGGCTCGCGCGCCCGCGAAGGGGACGCAGCGAAACCGGGGCCCGCGCCAGGCCAGCCGGGACGGACGCCGATGCCCGGGGCTGC"| __truncated__ "AGGGGACGCAGCGAAACCGGGGCCCGCGCCAGGCCAGCCGGGACGGACGCCGATGCCCGGGGCTGCGACGGCTGCAGAGCGAGCTGCCCTCGGAGGCCGGCGTGGGGAAGA"| __truncated__ "AGGGGACGCAGCGAAACCGGGGCCCGCGCCAGGCCAGCCGGGACGGACGCCGATGCCCGGGGCTGCGACGGCTGCAGAGCGAGCTGCCCTCGGAGGCCGGCGTGGGGAAGA"| __truncated__ ...
## $ xrefs :List of 14
## $ tfbs :List of 14
## $ exons :List of 14
## $ annotationFlags :List of 14
getRegion
# making a query through cbRegion to get all the clinically relevant variants
# in a specific region
library(cellbaseR)
cb <- CellBaseR()
res <- getRegion(object=cb,ids="17:1000000-1005000",
resource="clinical")
# to get all conservation data in this region
res <- getRegion(object=cb,ids="17:1000000-1005000",
resource="conservation")
#likewise to get all the regulatory data for the same region
res <- getRegion(object=cb,ids="17:1000000-1005000", resource="regulatory")
str(res,1)
## 'data.frame': 60 obs. of 12 variables:
## $ chromosome : chr "17" "17" "17" "17" ...
## $ source : chr "ccat_histone" "SWEmbl_R0005_IDR" "SWEmbl_R0005_IDR" "SWEmbl_R0005_IDR" ...
## $ featureType : chr "H3K27_trimethylation_site" "H3K27_acylation_site" "H3K4_monomethylation_site" "H3K9_acetylation_site" ...
## $ start : int 993140 997759 997760 998344 998400 999107 999880 997760 998344 998400 ...
## $ end : int 1004420 1000704 1002709 1000418 1000080 1000583 1000680 1002709 1000418 1000080 ...
## $ score : chr "." "." "." "." ...
## $ strand : chr "." "." "." "." ...
## $ frame : chr "." "." "." "." ...
## $ name : chr "H3K27me3" "H3K27ac" "H3K4me1" "H3K9ac" ...
## $ featureClass: chr "Histone" "Histone" "Histone" "Histone" ...
## $ alias : chr "K562_H3K27me3_ENCODE_YALE_ccat_histone" "Monocytes-CD14+_H3K27ac_ENCODE_Broad_SWEmbl_R0005_IDR" "Monocytes-CD14+_H3K4me1_ENCODE_Broad_SWEmbl_R0005_IDR" "Monocytes-CD14+_H3K9ac_ENCODE_Broad_SWEmbl_R0005_IDR" ...
## $ cellTypes :List of 60
getVariant
Getting annotation for a specific variant
library(cellbaseR)
cb <- CellBaseR()
res2 <- getVariant(object=cb, ids="1:169549811:A:G", resource="annotation")
# to get the data
res2 <- cbData(res2)
str(res2, 1)
A very powerfull feature of cellbaseR is ability to fetch a wealth of clinical data, as well as abiliy to fiter these data by gene, phenotype, rs, etc…
getClinical
library(cellbaseR)
cb <- CellBaseR()
# First we have to specify aour param, we do that by creating an object of
# class CellbaseParam
cbparam <- CellBaseParam(feature = "TET1", assembly = "GRCh38", limit = 50)
cbparam
## |An object of class CellBaseParam
## |use this object to control what results are returned from the CellBaseR
## methods
# Note that cbClinical does NOT require any Ids to be passed, only the param
# and of course the CellbaseQuery object
res <- getClinical(object=cb,param=cbparam)
str(res,1)
## 'data.frame': 50 obs. of 16 variables:
## $ chromosome : chr "10" "10" "10" "10" ...
## $ start : int 68574062 68574076 68574087 68574088 68574093 68574101 68574101 68574102 68574106 68574107 ...
## $ reference : chr "C" "C" "G" "G" ...
## $ alternate : chr "T" "A" "C" "C" ...
## $ hgvs :List of 50
## $ displayConsequenceType: chr "missense_variant" "missense_variant" "missense_variant" "missense_variant" ...
## $ consequenceTypes :List of 50
## $ conservation :List of 50
## $ geneExpression :List of 50
## $ geneTraitAssociation :List of 50
## $ geneDrugInteraction :List of 50
## $ traitAssociation :List of 50
## $ cytoband :List of 50
## $ id : chr NA NA "rs776152780" NA ...
## $ populationFrequencies :List of 50
## $ repeat :List of 50
cellbaseR wrappers
cellbaseE package also provides many wrapper functions around commomn cellbaseR
queries. These include:
- getClinicalByGene
- getTranscriptByGene
- getGeneInfo
- getSnpByGene
- getProteinInfo
- getClinicalByRegion
- getConservationByRegion
- getRegulatoryByRegion
- getTfbsByRegion
- getCaddScores
- getVariantAnnotation
CellbaseR utilities
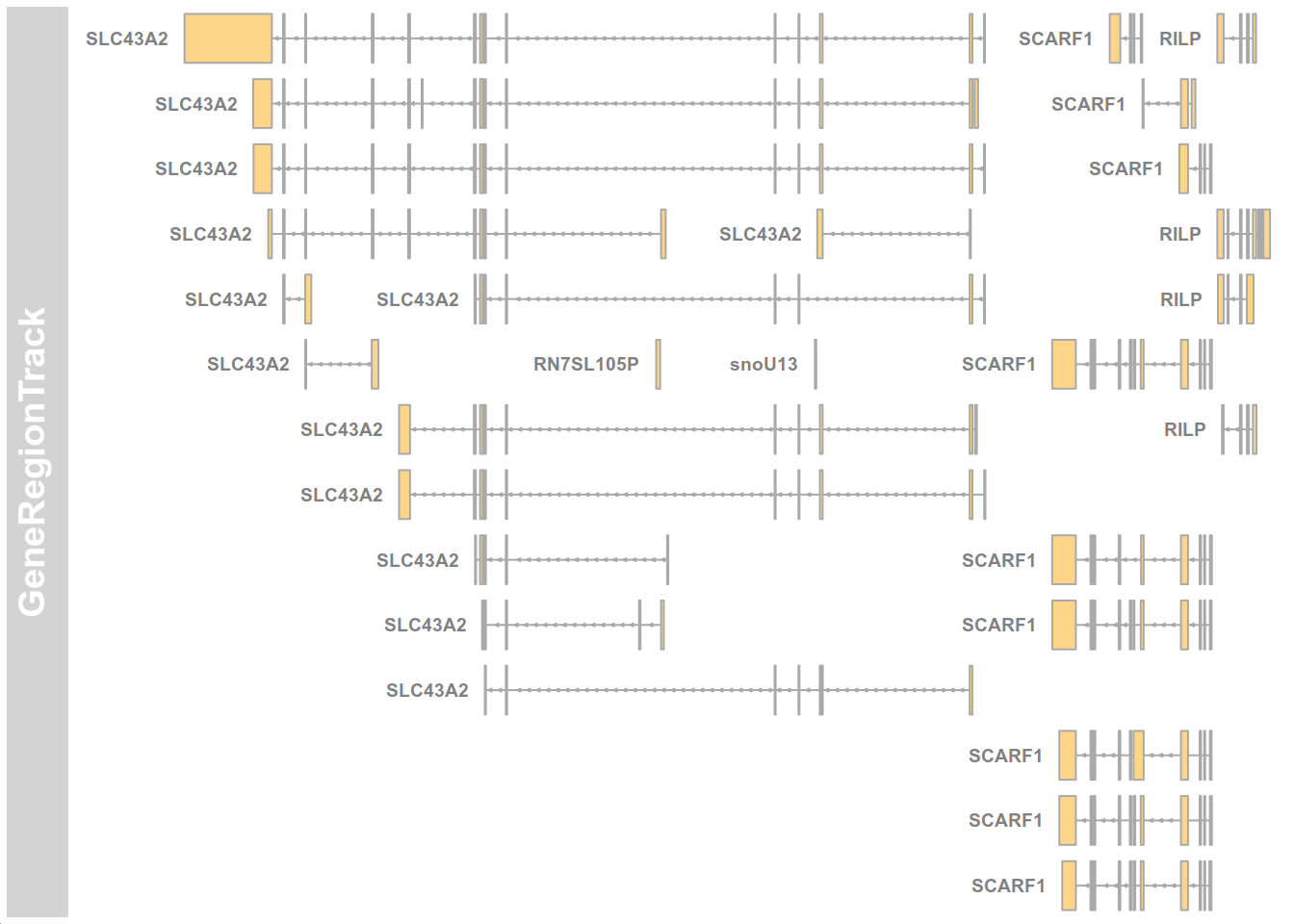
CreateGeneModel
A usefull utility for fast building of gene models, compatible with Gviz package for genomic visualization
library(cellbaseR)
cb <- CellBaseR()
test <- createGeneModel(object = cb, region = "17:1500000-1550000")
if(require("Gviz")){
testTrack <- Gviz::GeneRegionTrack(test)
Gviz::plotTracks(testTrack, transcriptAnnotation='symbol')
}
AnnotateVcf
This utility allows users to annoate genomic variants from small to medium-sized vcf files directly from the cellbase server, with a wealth of genomic annotations including riche clinical annotations like clinvar, gwas, and cosmic data, as well as conservation and functional scores like phast and cadd scores , without the need to download any files.
library(cellbaseR)
library(VariantAnnotation)
cb <- CellBaseR()
fl <- system.file("extdata", "hapmap_exome_chr22_200.vcf.gz",package = "cellbaseR" )
res <- AnnotateVcf(object=cb, file=fl, BPPARAM = bpparam(workers=2),batch_size = 100)
vcf <- readVcf(fl, "hg19")
samples(header(vcf))
## [1] "NA07034@1099927558" "NA07048@1099927687" "NA07055@1099927615"
## [4] "NA10846@1099927836" "NA10847@1099927741" "NA12146@1099927743"
## [7] "NA12239@1099927424" "NA12877@1099925716" "NA12878@1099927697"
## [10] "NA12891@1099927856" "NA12892@1099927810" "NA18503@1099927775"
## [13] "NA18504@0178873056" "NA18505@1099927412" "NA18506@1099927650"
## [16] "NA18524@1099927756" "NA18532@1099927601" "NA18912@1099927835"
## [19] "NA18913@1099927630" "NA18914@0178874379" "NA18940@1099927867"
## [22] "NA18947@0178875080"
length(rowRanges(vcf))==nrow(res)
## [1] TRUE
str(res,1)
## 'data.frame': 200 obs. of 18 variables:
## $ chromosome : chr "22" "22" "22" "22" ...
## $ start : int 16157603 17060707 17072347 17177682 17265124 17326914 17446991 17469049 17602839 17603503 ...
## $ reference : chr "G" "G" "C" "C" ...
## $ alternate : chr "C" "A" "T" "A" ...
## $ id : chr "rs370790235" "rs117836313" "rs139948519" "rs3020747" ...
## $ hgvs :List of 200
## $ displayConsequenceType : chr "intron_variant" "2KB_downstream_variant" "stop_gained" "non_coding_transcript_exon_variant" ...
## $ consequenceTypes :List of 200
## $ populationFrequencies :List of 200
## $ conservation :List of 200
## $ geneExpression :List of 200
## $ geneTraitAssociation :List of 200
## $ geneDrugInteraction :List of 200
## $ functionalScore :List of 200
## $ cytoband :List of 200
## $ repeat :List of 200
## $ variantTraitAssociation:'data.frame': 200 obs. of 2 variables:
## $ traitAssociation :List of 200
Mohammed O.E Abdallah
Post-Doctoral Fellow, Department of Human Genetics
My research interests include genomics data science, rare genetics disoders and scientific computing.